
bgchm: R package for hierarchical Bayesian genomic cline models with HMC. 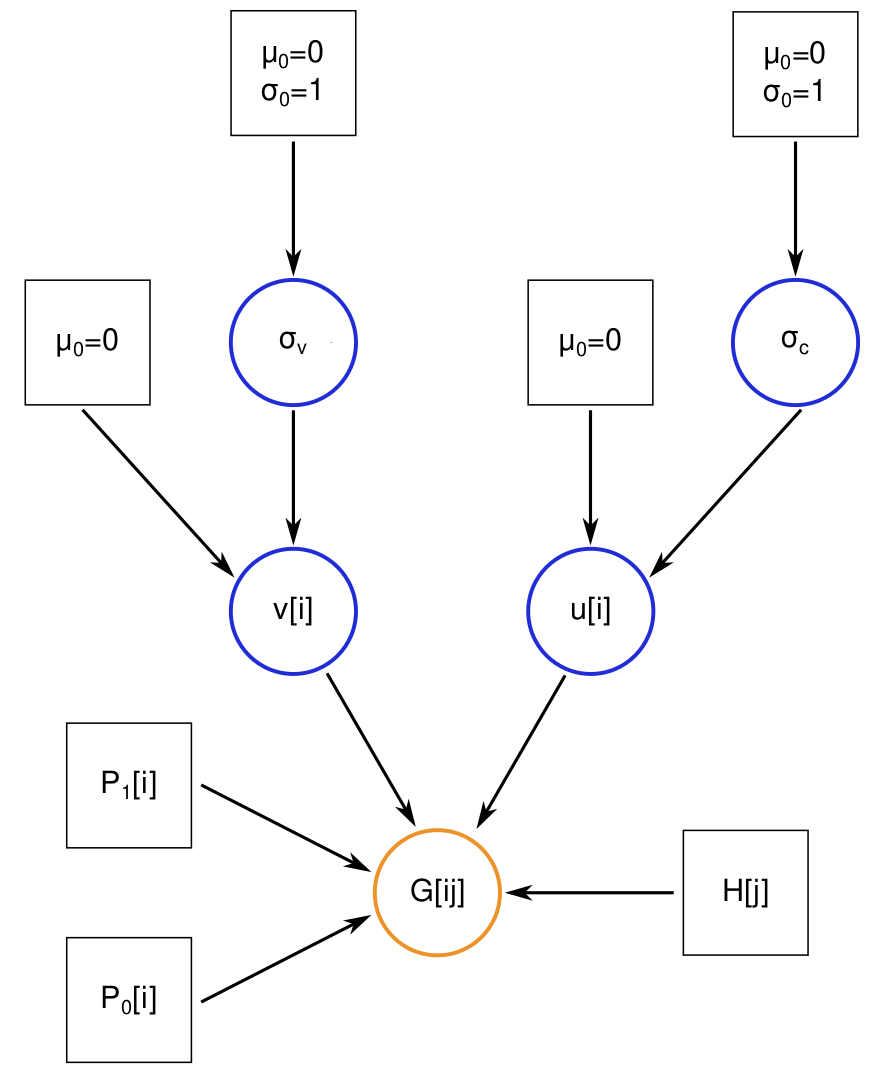 This is an R package for Bayesian analyses of population genomic data from hybrid zones, including Bayesian genomic cline analysis, estimation of hybrid indexes and ancestry class proportions, some geographic cline analyses, and accessory plotting functions. This package using Hamiltonian Monte Carlo (HMC) for sampling posterior distributions, with HMC sampling implemented via Stan.
This is an R package for Bayesian analyses of population genomic data from hybrid zones, including Bayesian genomic cline analysis, estimation of hybrid indexes and ancestry class proportions, some geographic cline analyses, and accessory plotting functions. This package using Hamiltonian Monte Carlo (HMC) for sampling posterior distributions, with HMC sampling implemented via Stan.
bgc: software for Bayesian estimation of genomic clines.  This software implements Bayesian estimation of genomic clines and can be used to estimate hybrid index and quantify genome-wide variation in introgression. Locus-specific measures of introgression can be used to study the genetics of adaptation and speciation. This software is compatible with traditional genetic data and next-generation sequence data with genotype uncertainty. An updated version of this program will be available soon!
This software implements Bayesian estimation of genomic clines and can be used to estimate hybrid index and quantify genome-wide variation in introgression. Locus-specific measures of introgression can be used to study the genetics of adaptation and speciation. This software is compatible with traditional genetic data and next-generation sequence data with genotype uncertainty. An updated version of this program will be available soon!
gbs2ploidy: software for inference of ploidy from GBS data. This R package contains functions for inference of ploidy from GBS data, including a function to infer allelic ratios and allelic proportions in a Bayesian framework. A paper describing the methods implemented in this software package is currently under review. E-mail me if you want the current draft. The package will also be available from CRAN.
introgress: software for analysis of introgression.  This R software package provides functions for quantifying introgression between hybridizing populations. This includes a likelihood-based method for estimating genomic clines using multilocus data and testing for deviations from null expectations. This software also implements a maximum likelihood estimate of hybrid index and provides several graphical summaries of genome-wide introgression.
This R software package provides functions for quantifying introgression between hybridizing populations. This includes a likelihood-based method for estimating genomic clines using multilocus data and testing for deviations from null expectations. This software also implements a maximum likelihood estimate of hybrid index and provides several graphical summaries of genome-wide introgression.
spatpg: software to infer selection from genetic time series.  This program can be used to estimate variance effective population size and environment-dependent selection from genetic time-series data (allele frequencies) from multiple populations. Selection is allowed to vary among populations and from generation to generation. Inference is made in a Bayesian framework.
This program can be used to estimate variance effective population size and environment-dependent selection from genetic time-series data (allele frequencies) from multiple populations. Selection is allowed to vary among populations and from generation to generation. Inference is made in a Bayesian framework.

fsabc: software to quantify polygenic selection from genetic time series. This program can be used to estimate the strength and variability of selection on traits and genetic variants in cases where selection varies by the environment and the trait under selection is polygenic. It requires a population-genomic time series, genotype-phenotype map, and data on the hypothesized environmental covariate. Inference uses approximate Bayesian computation.

Pingback: New software and paper | The Gompert Lab
Pingback: New method and software to measure fluctuating selection on polygenic traits | The Gompert Lab